视神经脊髓炎(Neuromyelitis optica,NMO),是一种罕见的自身免疫性炎性中枢神经系统脱髓鞘疾病,主要影响脊髓和视神经。Katherine Sterner 等人近日在 ACR 发布一例 60 岁女性视神经脊髓炎患者,并对该病进行总结,现翻录如下供大家参考学习。
病史
60 岁女性,左眼视力逐渐下降,双手感觉异常及体重减轻。发病前身体健康。
影像学表现
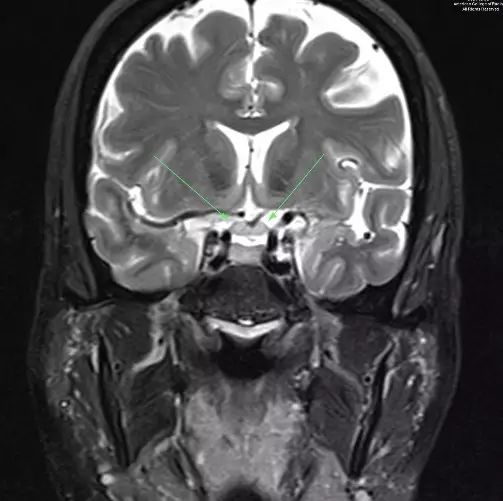
图 1. 冠状T2示视神经增粗(箭头示)
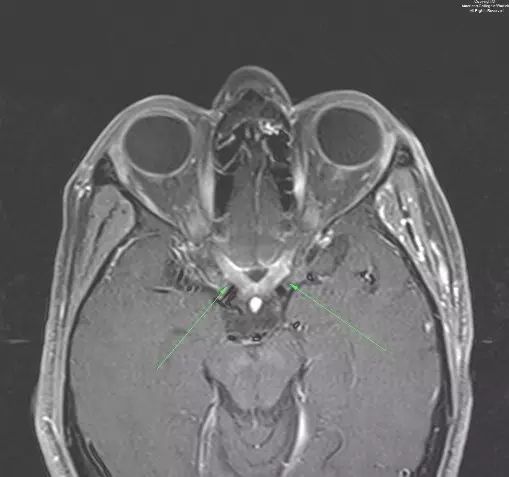
图 2. 增强T1示视交叉增粗并强化(箭头示)
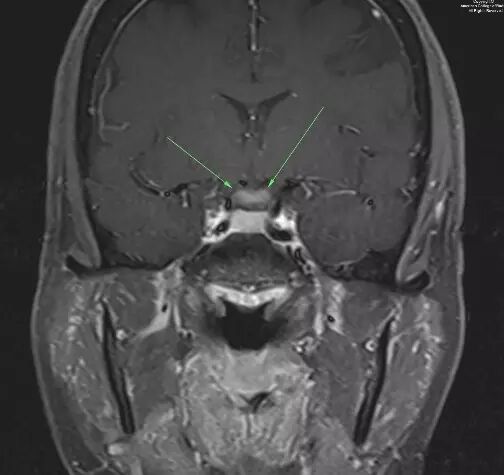
图 3. 冠状增强T1视交叉增粗并强化(箭头示)
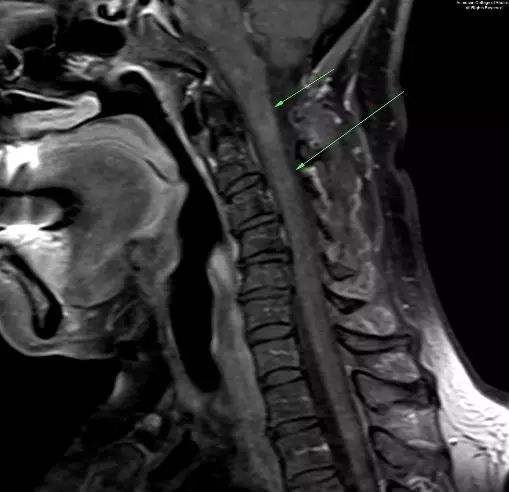
图 4. 矢状增强T1示颈椎内异常强化灶,范围从C1水平到C4下方(箭头示)
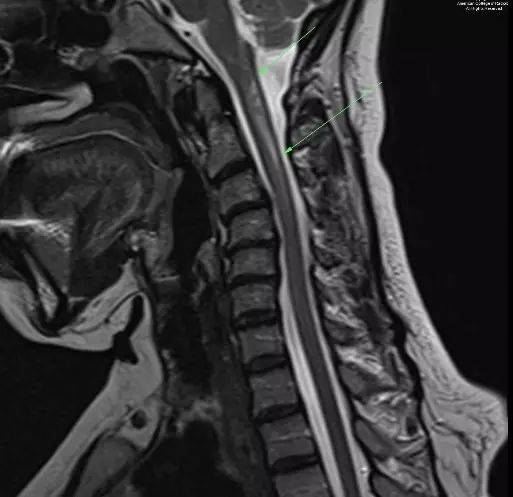
图 5. 矢状T2示C1到C4下方水平颈椎内脱髓鞘病灶(箭头示)
诊断
视神经脊髓炎
鉴别诊断
多发性硬化等疾病。
要点总结
视神经脊髓炎(Neuromyelitis optica,NMO),又叫 Devic 综合征,是一种自身免疫性炎性中枢神经系统脱髓鞘疾病,主要影响脊髓和视神经。在过去十年里,发现了一种名为水通道蛋白 -4 抗体 / 视神经脊髓炎特异性抗体(aquaporin-4 antibody/NMO-IgG)。这种抗体有助于将视神经脊髓炎与多发性硬化和其他脱髓鞘性疾病区分开。早期诊断 NMO 非常重要,因为一些用于其他类似脱髓鞘疾病(如多发性硬化)的药物可以加重 NMO 患者的病情。
病例分析
视神经脊髓炎,其主要特点是长节段横贯性脊髓炎和视神经炎同时出现。该病最早在 1870 年由 Sir Thomas Allbutt 报道。但该病却以Eugène Devic 的姓氏命名,这是因为 Eugène Devic 在 1894 年创造了「视神经脊髓炎」这一名词,并将该病描述为一种严重的临床变异型多发性硬化。
然而在 2004 年,随着一个新的血清学标志物的发现,即水通道蛋白 4 抗体(AQP-4 或神经脊髓炎特异性抗体 NMO-IgG),NMO 现在被认为是一个独立的疾病谱。
NMO 是一种罕见病,发病率约十万分之 4.4,且女性多于男性。发病年龄一般在 39 岁左右,晚于多发性硬化。该病的发病机制目前仍未明确,通常认为该病包括星形胶质细胞损伤、脱髓鞘、神经元丢失和坏死。MRI 检查可见受累视神经增粗以及 T2 信号增高,脊髓病灶呈长节段横贯性 T2 高信号。增强扫描病灶呈斑片状不均匀性强化,偶可见中央坏死和空洞形成。还可见大脑内的病灶呈 T2flair 高信号,这和多发性硬化的典型表现不同。
NMO 的诊断标准最初是由Dean M. Wingerchuk 在 1999 年制定的。2006 年,由于新的血清学标志物的发现,专家对诊断标准进行了更新,以更好的对该疾病谱进行描述。新的诊断标准将 AQP-4 血清抗体阳性作为支持性诊断标准。除了发作性视神经炎、横贯性脊髓炎之外,还需要 3 个支持性诊断条件中至少 2 个同时存在,才可以作出阳性诊断。
这 3 个支持性诊断条件包括:
(1)AQP-4 血清抗体阳;
(2)MRI 上脊髓连续病变延伸超过三个椎体节段;
(3)脑 MRI 表现与多发性硬化的表现不一致。
如今,NMO 抗体血清学阳性同时伴有视神经炎或横断性脊髓炎或有任何其他视神经或脊髓外的症状的患者均包括在 NMO 疾病谱内,并可以接受相应的治疗。同时更加强调了长节段横贯性脊髓病灶和 AQP-4 阳性在诊断该病时的特异性。如今 AQP-4 化验已经被广泛使用,其特异性接近 100%,但敏感性不稳定,从 30-80% 不等。因此,NMO 患者的 AQP-4 也可以为阴性,仅表现为视神经炎和横贯性脊髓炎。尚不清楚血清 AQP-4 情况是否会对复发率以及残疾预后有影响。
为了尽早进行治疗,早期鉴别 NMO 和多发性硬化非常重要。一旦误诊,一些药物如干扰素,可以加重 NMO 患者的病情。但是,和其他自身免疫性疾病类似,该病治疗的重点是急性发作后的快速恢复,以及防止复发。由于该病的罕见性,还没有太多随机对照临床试验来验证每种治疗方案的有效性。
一般情况下,对于急性发作性 NMO 可以使用大剂量的类固醇,并缓慢减量。如果症状恶化,可以使用血浆置换疗法。因为不完全恢复者有很高的复发率以及神经功能的累积性损伤,因此长期免疫抑制很重要。硫唑嘌呤或利妥昔单抗是一线治疗药物。如果上述药物副作用非常明显或治疗反应差,那么可以考虑霉酚酸酯,米托蒽醌,或甲氨蝶呤。 |









